Высокий рост, худоба, длинные пальцы и снижение зрения могут свидетельствовать о наличии опасного заболевания, с которым редко можно прожить больше 50 лет. Речь идет о генетически обусловленном синдроме Марфана (СМ). Им болел известный политический деятель Авраам Линкольн, эмир Усама Бен Ладен, а также другие исторические личности.
Содержание
- Что такое синдром Марфана?
- Общие сведения
- Патогенез
- Причины, факторы риска
- Классификация
- Код МКБ-10
- Симптомы
- Диагностика
- Лабораторное обследование
- Инструментальное обследование
- Лечение
- Питание
- Народная медицина
- Физиотерапия
- Последствия и осложнения
- Прогноз и продолжительность жизни
- Профилактика
- Известные личности с синдромом Марфана
- Заключение
Что такое синдром Марфана?
Синдром Марфана – это врожденная генетическая патология, характеризующаяся поражением соединительной ткани вследствие мутирования гена, отвечающего за кодирование коллагена. Проявляется различными скелетными, сердечно-сосудистыми отклонениями, зрительными расстройствами, проблемами с суставами.
У людей с синдромом наблюдается гигантизм (чрезмерно большой рост), аневризма аорты, арахнодактилия (паучьи пальцы), близорукость, эктопия хрусталика (смещение), плоскостопие, деформация грудной клетки, повышенная гибкость суставов, прочие аномалии.

Общие сведения
Синдром Марфана наследуется чаще остальных различных коллагенопатий. Однако все равно остается довольно редким недугом. Эта наследственная патология, диагностируется у 1 человека из 10-20 тысяч, независимо от расовой принадлежности или пола. Это довольно малый процент. Вероятность, что от больного родителя недуг будет наследоваться ребенку, составляет 50:50.
Патогенез
Больше половины человеческого веса представлено соединительной тканью. Из нее состоит скелет, кожа, сосудистые стенки.
Если смотреть более детально механизм возникновения заболевания, то нужно начать с того, что в каждой клетке тела, имеющей ядро, находятся 23 пары хромосом. Абсолютно каждая из хромосом была сформирована из 1 молекулы дезоксирибонуклеиновой кислоты (или ДНК). ДНК содержит большое количество генов. Так, каждая хромосома вмещает от 429 до 3511 генов.
Один из генов – фибриллин-1 находится на длинном плече 15-й хромосомы и обозначается как FBN1. Он отвечает за кодирование большого структурного белка, который входит в состав микрофибрилл (органеллы мышечных волокон) и отвечает за создание эластичности тканей, формирование прочного цитоскелета («скелет» клеточных структур). Дефекты фибрилина-1 приводят к мутации.
Эластиновые фибриллы – составляющая часть крупных сосудов и связок. Часто при нарушении со стороны этих фибрилл появляются аневризмы.
Синдром обуславливает поражение трансформирующего фактора роста бета (TGF-β), контролирующего пролиферацию (разрастание клеточных элементов) и клеточную дифференцировку (распределение их по функциям). Происходит нарушение связывания неактивной формы TGF-β, что повлечет за собой повышение активности фактора, а значит, проявятся все признаки синдрома.
По причине генетического нарушения происходит потеря эластичности кожи и другой соединительной ткани, потеря их прочности. Возникает разболтанность суставов, а кожа способна сильно растягиваться.
Ввиду изменения коллагеновых волокон происходит сбой нормального гемостаза, призванного сохранять жидкое состояние крови, растворять тромбы, останавливать кровотечения путем кровосвертываемости. Дефекты приводят к нарушению агрегации (объединения) тромбоцитов (один из элементов крови). Фибриллы (волокна цитоплазмы) участвуют в гемостазе. Наряду с замедленным кровотоком внутри сосудов происходит адгезия тромбоцитов к эластину (их прилипание) с помощью фибронектина. Фибронектин обеспечивает восстановление структур, создавая необходимые компоненты соединительной ткани – фибробласты. Лица, страдающие синдромом, имеют дефицит фибронектина.
Проблемы с ЖКТ обусловлены увеличенным содержанием в пищеварительной системе коллагена. Отмечается дисфункция билиарного тракта (желчевыводящие пути, желчный пузырь), хронический гастродуоденит, долихосигма (аномальное удлинение одного из отделов толстой кишки), грыжа пищеводного отверстия диафрагмы.
Вследствие вышеперечисленных факторов возникают характерные признаки синдрома.
Причины, факторы риска
Заболевание Марфана наследуется по аутосомно-доминантному типу. Мутация наследуется от родителя к ребенку по гену, лежащему в аутосомах (не половых хромосомах). Аутосомный тип значит, что проявление патологии никак не связано с полом, а доминантный тип означает, что мутация всегда появляется. По выраженности признаки варьируются ввиду наследственных особенностей. Определить аутосомно-доминантный тип можно:
- если заболевание наследуется каждым поколением семьи;
- соотношение здоровых и больных составляет 1:1;
- частота заболеваемости девочек и мальчиков одинакова;
- здоровые дети, у которых родители больны, имеют здоровых детей.
Основной причиной появления синдрома является мутация FBN1. Ген, отвечающий за продуцирование фибриллина-1 мутирует, а организм становится неустойчивым к различного рода деформациям, поскольку его структуры становятся сверх растяжимы. Фибриллин выступает важным белком межклеточного матрикса (вещество, заполняющее пространство внутри клеток или вне их), обеспечивающим сократимость и эластичность тканей.
Кроме того, что синдром наследуется, к мутации приводит также внешнее воздействие радиации, лучевой терапии.
Некая роль отводится нарушенному обмену веществ, вследствие которого в коллагеновых волокнах накапливается много мукополисахаридов. Впоследствии чего идет перерастяжение соединительной ткани.
Среди факторов риска можно отметить наследственность и возраст отца.
Синдром Марфана с большей долей вероятности будет наследоваться ребенку по мере увеличения возраста отца (шансы, что мутаген сможет наследоваться особенно значительные, если отцу больше 35 лет).
Страдающие болезнью с 50% долей вероятности будут иметь больных детей. Иногда о синдроме не догадываются. Можно привести пример ситуации.
Здорового парня возраста 16 лет, местную звезду баскетбола направили для генетического обследования по подозрению на синдром Марфана. Телосложение у парня такое же, как у его отца – субтильное. Отец мальчика умер внезапно во время пробежки. Другие члены семьи полностью здоровы, скелетных аномалий или зрительных расстройств не наблюдалось.
Медицинский осмотр показал, что у подростка астеническое телосложение (худощавость, удлиненные конечности, длинная шея, вытянутое лицо). Отмечается аркообразное нёбо, равное соотношение размаха рук к длине тела, деформированная грудная клетка, напоминающая «куриную грудку», арахнодактилия, стрии (кожные растяжки) на бедрах и плечах, диастолический шум. По результату эхокардиографии у парня расширен корень аорты, имеется аортальная регургитация (не смыкающиеся створки, приводящие к забросу крови к левому желудочку сердца). При офтальмологическом обследовании выявился двусторонний иридодонез (дрожание радужки во время выполнения резкого движения глазами), небольшой сдвиг хрусталиков кверху. Из этого следует вывод, что синдром Марфана диагностика подтвердила.
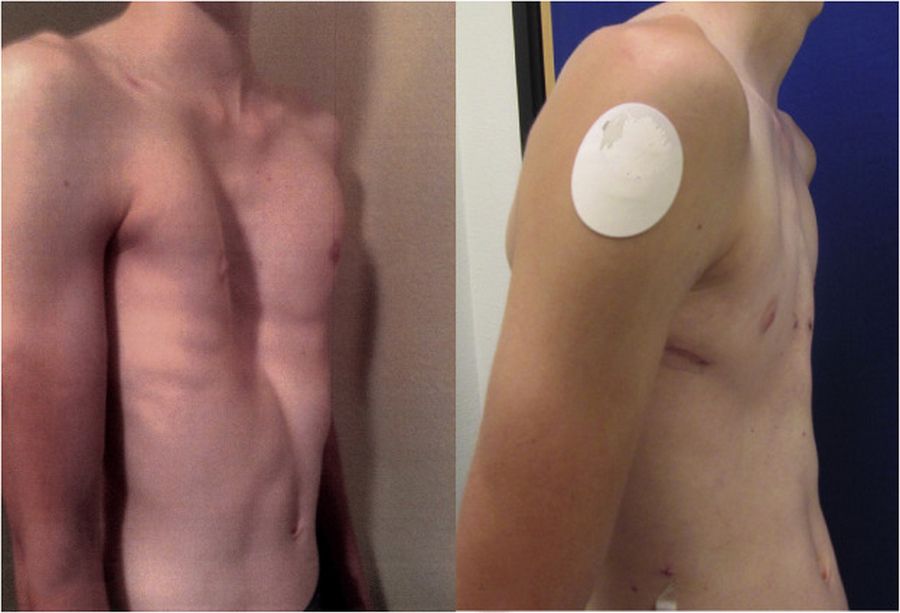
Деформация грудной клетки при синдроме
Классификация
Синдром Марфана протекает у всех по-разному. Учитывая степень тяжести, он бывает:
- легким;
- средним;
- тяжелым.
Тяжелые встречаются очень редко 1 к 50 000.
Исходя из того сколько структур организма поражено синдром Марфана классифицируется на 2 формы:
- стертую (присутствуют слабо выраженные отклонения в 1 или 2 системах);
- выраженную (присутствуют неявно выраженные отклонения в 3 системах или выраженные в 1; выраженные отклонения в 2, 3 системах или больше).
Характер течения бывает прогрессирующим (быстро прогрессирующие признаки, ухудшение состояния) и стабильным (неизменные признаки синдрома на протяжении многих лет).
Все случаи недуга подразделяются следующим образом:
- семейные (наследуется из поколения в поколение) – составляют 75%;
- случайные или спорадические (впервые возникшее в роду заболевание) – составляют 25%.
Код МКБ-10
Согласно международной классификации болезней 10-го пересмотра синдрому Марфана соответствует код Q87.4.
Симптомы
Для фенотипа страдающих данным заболеванием свойственна определенная протяженность: от легких вариантов дисплазии соединительной ткани до жизнеугрожающих случаев, связанных с серьезными системными расстройствами. Более распространен первый вариант.
Признаки синдрома чаще всего проявляются с возрастом, особенно дают о себе знать разнообразные скелетные аномалии во время развития костей (с раннего детства). У новорожденных заметны чересчур длинные пальцы, а остальные проявления развиваются позже, ближе к 7 годам. При рождении средний рост мальчика составляет порядка 53 см, а девочки – 52,5.
Признаки принято различать исходя из пораженной системы организма:
Опорно-двигательный аппарат
Ввиду наличия болезни (СМ) внешность человека выглядит специфически. У него наблюдается:
- гигантизм;
- деформированная килевидная грудная клетка (или как воронка);
- недостаточный вес;
- гипермобильность («разболтанность») суставов;
- короткое туловище;
- сколиоз (60% случаев), кифоз (горбатость), кифосколиоз;
- протрузии вертлужной впадины;
- удлиненные конечности (долихостеномелия);
- арахнодактилия;
- неловкость движений;
- ограниченность при разгибании локтевых суставов;
- спондилолистез (смещение позвонков относительно нижерасположенных);
- плоскостопие;
- мышечная гипотония (пониженный тонус мышц).
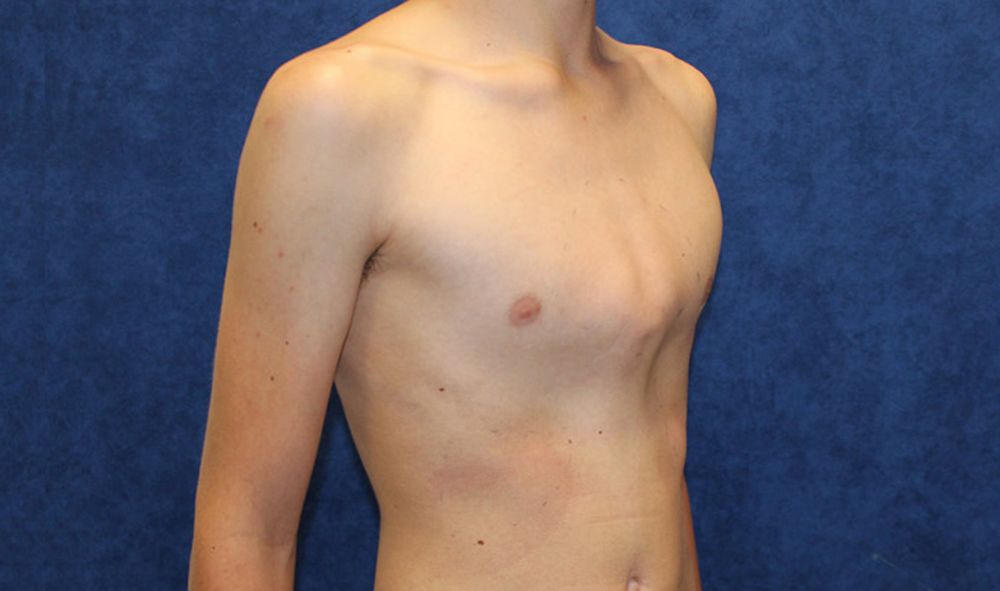
Килевидная грудная клетка
Вероятны черепно-лицевые аномалии – антимонглоидный разрез глаз, долихоцефалия (или длинноголовость), неправильный прикус, выпячивание верхней челюсти слегка вперед (прогнатия), ретрогнатия (сдвиг одной из челюстей назад), аркообразное нёбо, гипоплазия скуловых костей, энофтальм (глубокое расположение глаз в полости орбиты относительно нормы).
Сердечно-сосудистая система
Патологии сердца являются доминирующими среди всех симптомов, они зачастую определяют исход недуга. Эластичные стенки сосудов видоизменяются, растягиваются, чаще это происходит с аортой (аневризма) и крупными ветвями легочной артерии. Проявляются пороки сердца, его клапанного аппарата, перегородок. Восходящая часть аорты расширяется (дилатация), вовлекаются синусы Вальсальвы (пазухи), поражается митральный клапан (пролапс в 80% случаев).
При внутриутробном развитии у плода с имеющимся генетическим отклонением вероятно развитие таких пороков как, дефект межпредсердной перегородки, коарктация аорты, дефект межжелудочковой перегородки, сужение (стеноз) легочной артерии. Изменения со стороны сердца зачастую сопровождаются сбитым ритмом (тахикардия, фибрилляция предсердий), инфекционным эндокардитом (воспалением оболочки сердца). На ЭКГ наблюдаются признаки ИБС. Нередко у пациентов холодные руки, прослушиваются шумы в сердце, а возникающие кардиалгические боли отдают в верхние конечности, плечо, ключицу.
Нервная система
Психические процессы отличаются своеобразием. Люди с данным недугом отличаются повышенной раздражительностью, плаксивостью, завышенной самооценкой.
Синдрому свойственен значительный выброс адреналина, что способствует гиперактивности, нервной возбудимости, иногда появлению умственной одаренности, поэтому такие личности часто интеллектуально преуспевают.
Лишь в четверти случаев поступают жалобы на снижение интеллектуальных способностей, утрату радости, угнетение эмоционального фона. Часто проявляются нейроциркуляторная дистония (непереносимость стрессовых ситуаций, когда впоследствии возникает ощущение неполноты вдоха, «комок в горле»), депрессивные состояния. Утрачивается увлеченность профессиональной деятельностью, снижается социальная активность, чувство тревоги растет.
При ослаблении или растяжении дурального мешка (защитной пленки) позвоночника появляется поясничная, тазовая боль, болевые ощущения в ногах, цефалгия (головная боль). Возможно недержание мочи у детей. О поражении нервных структур свидетельствует ишемический или геморрагический инсульт, субарахноидальное кровоизлияние.
Орган зрения
Поскольку нарушается выработка фибриллина-1, то о нормальном функционировании цинновой связки, которая подвешивает хрусталик глаза, не может идти речь. Именно по причине ослабления этой связки появляется близорукость (миопия), подвывих хрусталика (или эктопия), что дальше чревато развитием глаукомы или полной слепоты. Возможны косоглазие, увеличение размеров роговицы, увеличение калибра сосудов глаз, гипоплазия (истончение) радужки. Иногда одним из проявлений являются голубые склеры. Реже встречается аниридия (отсутствие радужки), колобома (дефектное расщепление радужной оболочки).
Органы дыхания
У 5% пациентов возникает спонтанный пневмоторакс (накопление несвойственного для плевральной полости воздуха или газов). Встречается эмфизема легких (расширение воздушных пространств), гиперкапния (отравление переизбытком СО2, накапливающимся в крови). Присутствует кашель, одышка, поверхностное дыхание. Отягощающим обстоятельством становится дыхательная недостаточность. При рентгене иногда обнаруживаются апикальные псевдокисты.
Кожа, мягкие структуры организма
Для кожных покровов характерна излишняя растяжимость. Подкожная жировая клетчатка слабо развита. Это разновидность соединительной ткани, содержащая жировые отложения. Соединительнотканная недоразвитость обуславливает дефицит веса.
На коже образуются атрофические стрии (растяжки), чаще они заметны на плечах, груди, пояснице. Упомянутые стрии не связаны ни с беременностью, ни с набором или потерей веса. Ввиду ослабления соединительной ткани развивается варикоз. Поскольку подкожно-жировой слой недоразвит, случается грыжевое выпячивание передней брюшной стенки.
Органы пищеварения
Моторика билиарного тракта (желчный пузырь с протоками) нарушена. Возникает грыжа пищеводного отверстия диафрагмы, хронический гастродуоденит, дисбиоз (изменение состава микрофлоры).
Иногда среди отклонений из-за слабой соединительной ткани у пациентов встречаются грыжи (бедренные, паховые), разрыв связок, варикозное расширение вен, эктопия почек (смещение в нетипичное место, нередко при эктопии почки сморщиваются), опущение мочевого пузыря (цистоцеле) и матки, нефроптоз (опущение почек). Регистрируются кистозные новообразования в почках и печени, но они не считаются клинически значимыми.
Диагностика
При подозрении на синдром Марфана первым делом нужно обратиться к педиатру, он осмотрит пациента, затем даст направление к ортопеду, офтальмологу, генетику и кардиологу для подтверждения или опровержения диагноза и дальнейшей терапии в случае постановки диагноза.
При синдроме Марфана диагностика включает внешний осмотр пациента, сбор анамнеза (информацию об имеющихся других патологиях), лабораторные и инструментальные исследования.
Лабораторное обследование
Выполняется ДНК-анализ с мутирующим геном (автоматическое секвенирование). Обязательно назначают общий анализ крови, чтобы выявить признаки воспаления. Биохимический анализ крови показан тогда, когда нужно выявить дисфункцию определенного органа. Необходимо сдать анализ мочи для определения уровня гликозаминогликанов. Они показывают обмен веществ соединительной ткани. Обнаружение в моче оксипролина свидетельствует о тяжести протекания синдрома.
Инструментальное обследование
Аппаратная диагностика проводится, чтобы выявить дисфункцию внутренних органов и сосудистые нарушения. Инструментальные методики включают проведение:
ЭКГ (электрокардиография) – выполняется, чтобы обнаружить проблемы с сердцем или сосудами. При синдроме могут найти мерцательную аритмию, гипертрофию миокарда.
Эхокардиография – то же самое, что УЗИ сердца. Проводится для определения аневризм, увеличенной половины сердца.
Рентген – служит для обследования органов грудного отдела, просматривания изменений скелета.
Здоровье суставов и нервной системы проверяют с помощью компьютерной томографии или магнитно-резонансной томографии. Для отслеживания состояния аорты выполняют аортографию с введением контрастного вещества, которое окрасит нужные проблемные участки.
При зрительных расстройствах прибегают к офтальмоскопии (осмотра глазного дна).
При болезни Марфана диагностика упрощается тем, что пациенты имеют необыкновенную внешность. Они высокие, худые, с удлиненными конечностями. Характерные черты:
- маленький вес около 50 кг, при большом росте;
- длинные фаланги на руках;
- «птичье» выражение лица (небольшая челюсть, вытянутый череп, глубоко посаженные глаза);
- крупный нос;
- аркообразное нёбо.
Существуют способы, как определить синдром Марфана:
Признак первого пальца (симптом Штейнберга) – большой палец будет выраженно торчать наружу (за пределами гипотенара), если зажать руку в кулак.
Признак запястья (симптом Уолкера-Мердока) – при обхвате запястья первый палец сильно выходит за мизинец.
Пястный индекс – выполняется посредством рентгена. По снимку рассчитывают среднюю длину пясти. Берут значения ее длины, затем делят на ширину от второй до четвертой пястной кости. Должно получиться нормальное среднее значение – 5,4 – 7,9. При синдроме показатель выше 8,4.

Оценка вовлеченности соединительной ткани
| Признаки | Бал |
| Присутствующие признаки первого пальца (симптом Штейнберга) и запястья (симптом Уолкера-Мердока) | 3 |
| Присутствие одного из проявлений: симптома Штейнберга или Уолкера-Мердока | 1 |
| Килевидная грудная клетка | 2 |
| Асимметрия грудной клетки | 1 |
| Смещение медиальной лодыжки | 2 |
| Плоскостопие | 1 |
| Спонтанный пневмоторакс | 2 |
| Растяжение дурального мешка позвоночника | 2 |
| Протрузия (выступающая головка бедренной кости по направлению к малому тазу) вертлужной впадины | 2 |
| Уменьшенное соотношение верхней и нижней части туловища, увеличенное соотношение размаха рук к длине тела+ искривление позвоночника | 1 |
| Сколиоз, кифоз, кифосколиоз | 1 |
| Неразгибаемость локтевого сустава (угол менее 170°) | 1 |
| Наличие 3 черепно-лицевых аномалий (длинный череп, впалые глаза, маленькие скулы, ретрогнатия, антимонголоидный разрез глаз) | 1 |
| Стрии на коже (растяжки) | 1 |
| Стрии на коже (растяжки) | 1 |
| Близорукость (более 3 диоптрий) | 1 |
| Пролапс митрального клапана | 1 |
Диагноз подтверждается, если набирается больше 7 баллов.
Дифференциальную диагностику проводят с заболеваниями, подобными по симптоматике – синдром Шпринтцена-Гольдберга, MASS-синдром, синдром Элерса-Данло, пролапс митрального клапана, синдром Лойса-Дица.
Лечение
Поскольку синдром Марфана неизлечимое заболевание, лечение основано на симптоматической терапии и исправлении уже существующих дефектов. Следует поддерживать стабильное состояние с помощью медикаментов. Врачи назначают:
Бета-адреноблокаторы (обладают гипотензивным воздействием, уменьшают частоту сердечных сокращений, угнетают сократимость миокарда) – Атенолол, Анаприлин, Пропраналол.
Блокаторы кальциевых каналов (используются для коррекции тонуса сосудистых стенок) – Верапамил, Нифедипин. Эти препараты используются, если к бета-адреноблокаторам имеются противопоказания.
Ингибиторы АПФ (применяют при лечении повышенного артериального давления) – Каптоприл, Лизиноприл.
Метаболики (регулируют процессы обмена) – Милдронат, Рибоксин.
Препараты, стимулирующие и нормализующие коллагеновые волокна – Структум, Алфлутоп, Румалон.
Антиоксиданты (нейтрализующие свободные радикалы), Антигипоксанты (противостоят недостаточному снабжению кислородом тканей) – Элькар, Коэнзим Q10.
Антикоагулянты (против образования тромбов) – Гепарин.
Ноотропы (улучшают психические функции) – Винпоцетин, Пирацетам.
По необходимости врач назначит антибиотики, чтобы избежать инфекционного эндокардита.
Обязательно принимать поливитаминные комплексы.
Если зрение начало снижаться врач пропишет носить очки, либо линзы, чтобы офтальмологическая проблема не прогрессировала.
По показанию проводится хирургическое вмешательство. Оно направлено на исключение 3 основных проблемных факторов:
- Операция на сердце или крупных сосудах – реконструкция сердца, протезирование аорты, пластика клапанов сердца.
- Офтальмологическая операция – удаление глаукомы или катаракты, замена хрусталика, лазерная коррекция близорукости.
- Операция по исправлению осанки и суставов – пластика грудной клетки, эндопротезирование суставов, декомпрессия позвоночника, его стабилизация, выравнивание с применением металлических пластин.
Питание
Показана диета с продуктами, где содержание магния достаточно велико. За счет этого микроэлемента поврежденные сосуды быстрее восстанавливаются. Помимо магния стоит обогащать рацион белками, витаминами, жирными кислотами. С целью оздоровить сосуды следует ежедневно употреблять орехи, какао, сухофрукты, гречку, ячневую крупу.
Для прочности суставов, связочного аппарата и костей нужно употреблять натуральные хондропротекторы. Они не только участвуют в строительстве костных и хрящевых тканей, а еще нормализуют обмен веществ. Источником натуральных хондропротекторов являются хрящи животного происхождения, заливное желе, холодец, наваристые бульоны, красная рыба (семга, кета, лосось), мармелад.
Меню людей, болеющих синдромом Марфана, должно включать много растительной пищи (фрукты, овощи, зелень), морепродукты, крупы, растительное масло, печень, птицу, молочные продукты.
Из рациона необходимо исключить тугоплавкие сыры, копчености, алкоголь, кофе, газировку.
В крови страдающих синдромом часто повышен уровень соматотропного гормона, что обуславливает «избыточный рост», для его подавления с детства в рацион вводят высокожировые энпиты (сухие молочные продукты, где регулируется ценность и состав пищи), сокращающие продуцирование соматотропного гормона.
Народная медицина
Синдром Марфана наследуется генетически, а подобные болезни народными средствами излечить невозможно, но убрать или ослабить неприятные симптомы, сопровождающие синдром реально.
Если отмечается недостаточность соединительной ткани, то необходимо использовать растения, обладающие анальгезирующим эффектом, укрепляющие вены, улучшающие метаболизм. Среди них:
- донник;
- одуванчик;
- лопух;
- березовые листья;
- кора ивы;
- боярышник;
- девясил;
- сабельник болотный;
- конский каштан;
- клевер луговой.
Одну из трав заваривают на выбор, либо можно сочетать вместе несколько. В аптеке продаются готовые травяные сборы – Артрофлор или Ангиофлор.
Для повышения иммунитета показаны витаминизированные чаи, мумие, бракшун (или каменное масло), настойка восковой моли, отвары из календулы, череды или солодки.
При болях в суставах помогает медовый массаж или растирки. Используются смеси меда с алоэ или мед с фурацилином.
Физиотерапия
Физиотерапевтические процедуры показаны при стертой форме болезни. С раннего возраста желательно посещать курсы массажа, заниматься лечебной физической культурой (ЛФК). Зачастую упражнения проводят в лежачем положении во избежание перенагрузок на позвоночник.
Людям с данным синдромом следует пройти санаторно-курортное лечение. Принять углекисло-сероводородные ванны, пройти электрофорез с лекарственными препаратами, лечение ультразвуком. Полезными для позвоночника, суставов и мышц будут лечебные грязи.
Последствия и осложнения
По мере прогрессирования синдрома вероятность возникновения осложнения довольно высока. Самые частые осложнения это:
- катаракта (помутнение хрусталика);
- глаукома (периодическое повышение внутриглазного давления);
- полная слепота;
- нарушения осанки (последние степени кифоза, сколиоза);
- пролапс митрального клапана (провисание его створок, когда происходит сокращение левого желудочка сердца);
- застойная сердечная недостаточность, обусловленная нарушенной сократимостью сердечной мышцы;
- расслаивающая аневризма;
- спонтанный пневмоторакс;
- пневмония;
- инсульт;
- тромбозы.
Самое тяжелое осложнение – разрыв аневризмы, которое ведет к смерти.
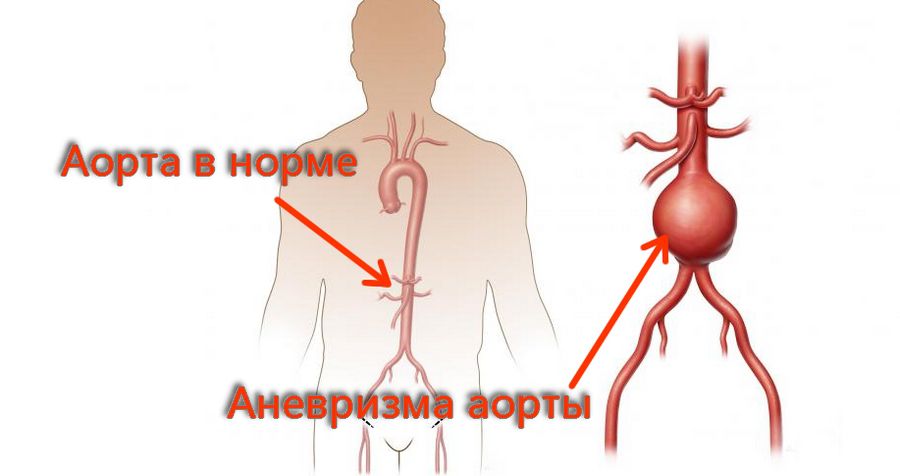
Наиболее частое осложнение синдрома, которое заканчивается смертельно
Прогноз и продолжительность жизни
Прогноз во многом зависит от степени сердечно-сосудистых и скелетных изменений, а также нарушений со стороны зрения. Если игнорировать лечение, то срок жизни редко превышает 30-45 лет, а ему грозит внезапная смерть, но при своевременной помощи прогноз более утешителен. Современная медицина добилась того, что больные доживают до преклонного возраста (до 70), при этом ведут умеренный образ жизни. Однако смертность все еще остается высокой (вероятность не дожить до 50 лет составляет 90-95%). Существует высокий риск возникновения осложнений. Инвалидность получают за снижение зрения.
Основной причиной смерти становится – сердечная недостаточность, спровоцированная аневризмой, последующим разрывом аорты и регургитации (обратный ток крови) сердечных клапанов. Данные события случаются при физических нагрузках. В этом кроется вся опасность.
Поскольку люди с этим синдромом отличаются астеническим телосложением, особенно длинными руками, это позволяет им пойти в спорт из-за широкого размаха рук, но как раз такие нагрузки несут смертельную опасность.
Профилактика
Какая-либо специфическая профилактика не разработана, поэтому предупредить мутацию, которая в половине случаев обязательно будет наследоваться никак нельзя, но для уже болеющих синдромом Марфана существует ряд ограничений, что предостережет их от тяжелых последствий.
Запрещены:
- сильные физические нагрузки;
- участие в спортивных соревнованиях, подводном плавании, контактных видах спорта;
- работы на вредном производстве;
- нахождения в месте, где радиационный фон высокий.
При возможности лучше сменить жаркий климат на умеренный.
Болеющим необходимо регулярно наблюдаться у врача (каждое полугодие). Разрешается легкая физическая нагрузка, спортивная ходьба, игры с мячом, элементарные упражнения (приседания, повороты, растяжка).
Не стоит игнорировать кариес, обычную простуду или глистные инвазии, так как они тоже значительно усугубляют течение синдрома, поскольку иммунитет при таком диагнозе намного слабее, чем у здоровых людей.
Для предупреждения болезни Марфана диагностика должна быть проведена еще на этапе зачатия, чтобы просчитать все риски, поскольку патология наследуется с высокой долей вероятности.
Известные личности с синдромом Марфана
Данной патологией страдали некоторые известные личности. Генетическая патология была у итальянского композитора и скрипача Никколо Паганини. Благодаря тонким, гибким пальцам он смог достичь успеха в музыкальной сфере. По описаниям Паганини был худощавым, бледным, с деформированной грудной клеткой и не ровными по длине ногами. Его манера исполнения запомнилась лихорадочными движениями смычка на концертах.
Ханс Кристиан Андерсен – писатель и сказочник родом из Дании. Среди его произведений самые известные «Русалочка», «Гадкий утенок», «Дюймовочка», «Снежная королева». У писателя из-за синдрома рано развились проблемы со зрением.
Авраам Линкольн (американский политик) тоже страдал этим недугом. Его рост составлял 193 см, наблюдалось непропорциональное телосложение, длинноватые конечности, «расшатанность» суставов, ревматические боли.
Русский писатель Корней Чуковский, как считают исследователи, имел генетический дефект. Медики обратили внимание на огромный непропорциональный лицу нос писателя, длинные ноги и руки.
Усама Бен Ладен – основатель террористической организации «Аль-Каида». Усама всегда носил при себе трость и предпочитал мешковатую одежду, чтобы скрыть очень малый вес (72 кг) против большого роста (195 см). У него наблюдались деформированные фаланги и вытянутое лицо, что свидетельствовало о наличии генетического дефекта.
Синдром был обнаружен у Майкла Фелпса (американского пловца), Джонатана Ларсона (американского композитора), Винсента Скьявелли (американского актера).
Заключение
Синдром Марфана – это неизлечимая болезнь, но при адекватном медицинском лечении удается добиться стабильной ремиссии, продлить срок жизни, при этом сохраняя право на полноценную жизнеспособность. К сожалению, наблюдаться у врачей придется пожизненно. Лицам с синдромом Марфана, чтобы не допустить рождения больного ребенка, следует перед зачатием пройти генетические обследования. При беременности необходима пренатальная диагностика, определяющая по УЗИ нормальное внутриутробное развитие плода, следует выполнить скрининг сывороточных маркеров. Стоит помнить о том, что синдром Марфана наследуется в 50% случаев.


