Случается так, что ребенок нормально развивается, учится ходить, разучивает новые слова, а затем эти приобретенные навыки начинают быстро исчезать, хотя внешне ничего не предвещало беды. В таком случае речь идет о синдроме Ретта.
Содержание
- Синдром Ретта – что это за болезнь
- Общие сведения
- Патогенез
- Причины
- Почему синдром диагностируется только у девочек
- Стадии
- МКБ-10
- Симптомы
- Диагностика
- Критерии синдрома Ретта
- Атипичные формы синдрома
- Лабораторная диагностика
- Инструментальная диагностика
- Дифференциальная диагностика
- Лечение
- Медикаментозное лечение
- Реабилитация
- Питание
- Последствия и осложнения
- Прогноз и продолжительность жизни
- Профилактика
- Заключение
Синдром Ретта – что это за болезнь
Синдром Ретта – это прогрессирующее генетическое заболевание, выявляющееся преимущественно у девочек (очень редко у мальчиков). При синдроме поражается центральная нервная система и ранее приобретенные навыки начинают исчезать. Пропадает способность контролировать движения рук, происходит частичная или полная утрата речи, замедляется рост головы. Болезнь характеризуется умственной отсталостью тяжелой степени. Наличие синдрома влияет на продолжительность жизни. С таким диагнозом долго не живут.

 |
Статья проверена медицинским экспертом Маслиховой Любовью Васильевной |
Общие сведения
Впервые о существовании этой патологии стало известно в 1954 г. Ее обнаружил австрийский невролог Андреас Ретт, но дал описание болезни в 1966-ом. А вот признали это заболевание аж в 1983 г. после того как про недуг опубликовал статью шведский исследователь Бенгт Хагберг.
Распространенность синдрома Ретта такова: болезнь встречается у 1 из 10-15 тыс. младенцев. Тип наследования – доминантный.
Географически болезнь распространена не равномерно. Всплеск характерен для мест небольшой популяции, где низкий процент людей, выходящих из разных антропологических групп, а также в Венгрии, Норвегии, Албании, Италии.
Патогенез
Науке известно 800 различных мелких мутаций, приводящих к синдрому. Если ген (МEСP2), находящийся на плече Х-хромосомы дефектен, то это приводит к боезни.
Ген МЕСР2, где происходит мутация, отвечает за кодирование метил-СpG-связывающего белка 2 (МеCр2). Больше всего этого белка содержится в нейронах (нервных клетках). Однако на начальных этапах формирования эмбриона этого белка мало, по мере взросления он увеличивается, а значит, риск возрастает. Если ген не поврежден, то на определенном этапе его белок отключает некоторые другие гены, и развитие мозговых структур происходит нормально. При мутации же никакого отключения не происходит, и мозг развивается неправильно.
Новорожденный выглядит нормально, идет обычное правильное формирование, но ближе к 6-18-месячному возрасту начинается частичный или полный регресс психического развития и приобретенных навыков. Наблюдается аномалия походки, присоединяются дыхательные расстройства, судороги, речевые проблемы.
Размеры мозга, у пациентов при наличии синдрома, маленькие, имеется атрофия коры и мозжечка, но разрушения нейронов нет. Это не типичная нейродегенеративная болезнь. Нейроны меньше по размеру, чем должны быть, они более плотно размещены, а ветвление дендритов (отростки от тел нейронов) упрощенное.
Причины
Примерно 99% случаев заболевания спонтанны, дефектный ген может появиться «из ниоткуда». Семейная передача синдрома Ретта встречается менее чем в 1%. Бывали случаи, когда недуг передавался от здоровой матери или матери, имеющей не полностью дефектный ген. Новые мутации в 70% случаев происходят из половых клеток отца.
Причиной деградации является мутация гена MЕСР2 на плече Х-хромосомы, отвечающего за синтез белка, который должен подавляться со временем, но из-за того, что ген поврежден, этого не происходит, впоследствии мозговые структуры развиваются неправильно.
Почему синдром диагностируется только у девочек
Каждые 90 минут на свет появляется новорожденный с синдромом Ретта. Страдают этим заболеванием лишь девочки.
По доминантному типу наследования болеть будут преимущественно девочки, поскольку мутирует ген (MЕCР2), находящийся на плече X-хромосомы. У девочек таких хромосом 2. У мальчиков нет парной Х-хромосомы (у них XY), поэтому любая мутация гена, связанного с ней заканчивается летальным исходом среди младенцев мужского пола обычно еще во внутриутробном периоде, поскольку нет одной запасной здоровой хромосомы. Чаще причиной смерти становится перинатальная энцефалопатия. Однако продолжительность жизни девочек тоже сокращена до 30, максимум до 50 лет.
Известны единичные случаи, когда у мальчиков обнаруживали такую патологию, но они выживали. При наличии у лиц мужского пола полисомии – аномалии, характеризующейся дополнительной хромосомой, определяющей женский половой признак (синдром Клайнфельтера) возможно возникновение синдрома Ретта, где она из них будет дефектной, а вторая здоровой.
Стадии
Насчитывается 4 стадии синдрома Ретта после того, как патология будет обнаружена:
Первая (начинается с 6 месяцев). Еще называется стадией стагнации. Длится 2-4 месяца. Останавливается психомоторное развитие, замедляется рост костей скелета, головы, стоп. Отмечается потеря социальной улыбки, пропадает интерес к играм. Наблюдается мышечная гипотония (пониженный мышечный тонус). Диагноз еще сложно установить.
Вторая (начинается с 1-3 лет). Называется стадией быстрого регресса (деструктивной). На второй стадии регистрируется убывание психомоторного развития, постепенная утрата навыков, появляются стереотипные двигательные акты, являющиеся показательными. Стереотипии выражаются постукиваниями ладошками по лбу, движениями наподобие «мытья рук», заламываниями, оскалом. Формирование лепета, экспрессивная речь теряется, характерно воспроизведение отдельных звуков. Периодические крики, проблемы с засыпанием.
Третья (начало с 3 до 10 лет). Псевдостационарная стадия. Наступает небольшое затишье синдрома, симптомы аутистического характера пропадают, сон нормализуется. Стереотипии верхних конечностей сохраняются, наблюдается расстройство со стороны гнозиса (способность к запоминанию предметов, их понимаю, восприятию) и праксиса (способность к выполнению ряда заученных действий). Вследствие нарушенного проведения нервного импульса возникает тремор (непроизвольное дрожание частей тела), динамическая или статическая атаксия (раскоординация), мышечная дистония (несогласованное сокращение мышц). Присоединяются эпилептические приступы, периоды гипервентиляции (избыток кислорода впоследствии интенсивного дыхания), обмороки.
Четвертая (начинается после 10 лет). Еще называется стадией поздних двигательных нарушений. Отмечаются ярко выраженные моторные расстройства: ригидность мышц нижних конечностей (неподатливость, оцепенение), усиление парезов (параличей), сколиоза, кифосколиоза. Ближе к подростковому возрасту утрачивается способность самостоятельно передвигаться.
Если недуг прогрессирует стремительно, это свидетельствует о высоком риске внезапной смерти.
МКБ-10
Согласно международному классификатору болезней 10-го пересмотра синдрому Ретта соответствует код F84.2.
Симптомы
Одними из самых ярко выраженных симптомов является психо-речевое отставание и двигательные расстройства.
Вначале младенец нормально развивается, учится говорить, выполнять целенаправленные манипуляции руками, ходить, но после 6 или 18 месяцев теряются все приобретенные ранее навыки. Деградация происходит в кратчайший период.
Наблюдается следующая клиническая картина:
- Частичная или полная утрата мелкой (тонкой) моторики (манипуляции пальцами).
- Утрата лепета или выученных слов (мутизм).
- Стереотипное движение рук перед собой (изображающее мытье, похлопывания, закладывание рук в рот).
- Нарушенные коммуникативные способности (избегание контактов с другими, отстраненность, потеря интереса к играм).
- Аутостимуляция (раскачивания, мычание, игры со своим языком).
- Замедленный рост головы.
Дополнительно могут появляться следующие признаки:
- Гипервентиляция, непроизвольная задержка дыхания (апноэ). Довольно распространенный симптом. Апоэ может продолжаться до 40 секунд, а поскольку дыхание связано с сердечным ритмом, то это вредит сердцу. Возможно возникновение кислородного голодания сердечной мышцы. Гипервентиляция же проявляется интенсивностью дыхания, что повышает потребность организма в кислороде.
- Заглатывание лишнего воздуха при употреблении пищи. Иногда чрезмерное накопление воздуха вызывает ярко выраженное вздутие живота (у 5%).
- Бруксизм (скрежет зубами). Случается по причине непроизвольного сжатия челюстей.
- Диспраксия (неуклюжесть), апраксическая походка (слабость в ногах отсутствует, но сложно оторвать стопы от пола, получается шаркающая походка).
- Искривление спины. В раннем школьном возрасте возможна деформации позвоночника. При чрезмерном искривлении требуется оперативное вмешательство.
- Неправильное положение ног. С наступлением школьного возраста у детей наблюдается деформация положения стоп, что затрудняет устойчивость и ходьбу. Деформации ассиметричны, более выражены справа.
- Гипопластичность ступней. Заметно, что ступни непропорционально маленькие. Вследствие нарушенного питания тканей в подростковом периоде, кожа становится синюшной, а ноги холодными.
- Эпилептические приступы (фокальная эпилепсия).
- Неадекватный смех посреди ночи или пронзительные крики. Ребенок начинает смеяться ночью, причем смех продолжается несколько часов. У подростков наблюдаются продолжительные пронзительные крики. Исследования показывают, что они не обусловлены болью, а являются следствием дисфункции головного мозга.
- Проблемы со сном.
- Отсроченная реакция на боль. Реакция обычно держится довольно длительно.
- Общение посредством взгляда. Чтобы компенсировать отсутствие речи или жестикуляции у детей развивается коммуникация через взгляд. Они смотрят на объекты, на которые хотят обратить внимание окружающих.
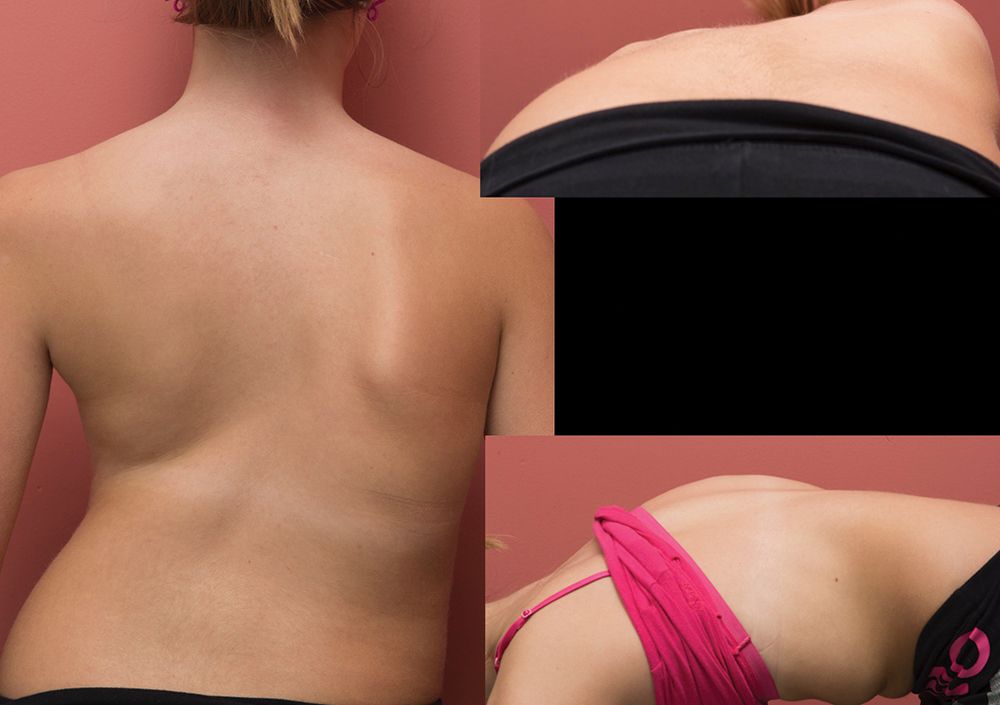
Искривление позвоночника при синдроме Ретта
Помимо типичных признаков отмечаются трудности питания. Вследствие таких факторов, как непроизвольное поворачивание языка, трудное пережевывание и проблемы с глотанием, процесс трапезы значительно замедляется, а вес снижается.
Происходит сбой регуляции тонуса мышечных стенок ЖКТ, что способствует рвоте, образованию запоров или обратному забрасыванию пищи в пищевод из желудка (гастроэзофагеальный рефлюкс). Вследствие этих проявлений у малыша возникает страх и отвращение к трапезе.
Диагностика
Диагностикой синдрома Ретта занимается педиатр-неонатолог или детский психиатр. Потребуется также помощь невролога, мануального терапевта.
Обязательно сначала проводят экспериментально-психологическое исследование (ЭПИ), подразумевающее анализ личности, особенности и психическое нездоровье.
Критерии синдрома Ретта
Существуют диагностические критерии, по которым определяется болезнь, симптомы таковы:
| Необходимый критерий | Дополнительный критерий | Исключение |
| • перинатальный период протекает нормально; • первые 6 месяцев младенца психомоторика развивается нормально; • нормальный размер окружности головы у новорожденного; • задержка роста головы с пятимесячного возраста; • утрата нормальной речи, намеренных двигательных актов верхних конечностей (6 мес. – 2,5 лет); • тяжелая умственная отсталость; • стереотипные движения: заламывание рук, гримасничанье, скрежет зубами, хлопки; • атаксия (несогласованность двигательных действий), апраксия (непоследовательность действий) туловища. |
• отставание в росте; • эпилепсия; • спастичность (повышенный мышечный тонус); • кифосколиоз; • маленькие стопы; • нарушенное дыхание; • вазомоторные нарушения (жар, потливость, головокружения); • аномалии при ЭЭГ; • периферические расстройства (невропатии). |
• ретинопатия (поражение сетчатки глаза); • внутриутробная задержка роста; • микроцефалия (уменьшенный размер черепа); • органомегалия (увеличение органов); • перинатальная энцефалопатия (повреждения головного мозга на 28-й неделе беременности); • неврологические нарушения на фоне травмы или инфекции; • наличие прогрессирующей патологии. |
Атипичные формы синдрома
Атипичная форма проявляет себя иначе:
| Основной критерий | Дополнительный критерий |
| • частичное или полное отсутствие речевых навыков; • частичное или полное отсутствие коммуникативного умения; • сниженная скорость роста головы у годовалых малышей; • периодичность течения болезни; • постоянные однообразные стереотипии верхних конечностей. |
• нарушенное дыхание; • нарушение локомоции (передвижения в пространстве); • аэрофагия (заглатывание дополнительного воздуха при поедании пищи); • амиотрофия ног (нарушенная трофика); • плохой зрительный или эмоциональный контакт; • сниженная реакция на боль; • бруксизм (зубной скрежет); • голосовые стереотипии. |
Самой распространенной атипичной формой является мозаичная, при которой тяжесть патологии зависит не только от степени, а и от формы. Синдром иногда протекает атипично:
С сохраненной речью – присутствуют неярко выраженные проявления. Умение говорить частично сохраняется (хотя словарный запас небольшой), физическое развитие нормальное, средне-тяжелое слабоумие, умеренный сколиоз. Девочка способна воспроизводить от 20 слов до несколько лишь сотен или же говорить длинными грамотными предложениями, задавать одни и те же вопросы, что позволяет спутать синдром с аутизмом.
Вариант Rolando – наблюдается сильно выраженная задержка психомоторики, двигательная стереотипия, дыхательные нарушения, теряется способность к самостоятельному передвижению.
Вариант Hanefeld – рано развиваются судорожные приступы, еще до умственной деградации.
Лабораторная диагностика
Чтобы выявить пораженный ген, нужно провести молекулярно-генетический анализ. Изучать наследственный анамнез не целесообразно, так как мутации спонтанны. Наиболее информативным будет прямое секвенирование.
Кроме того пациенту нужно будет сдать общий анализ крови (ОАК) и общий анализ мочи (ОАМ).
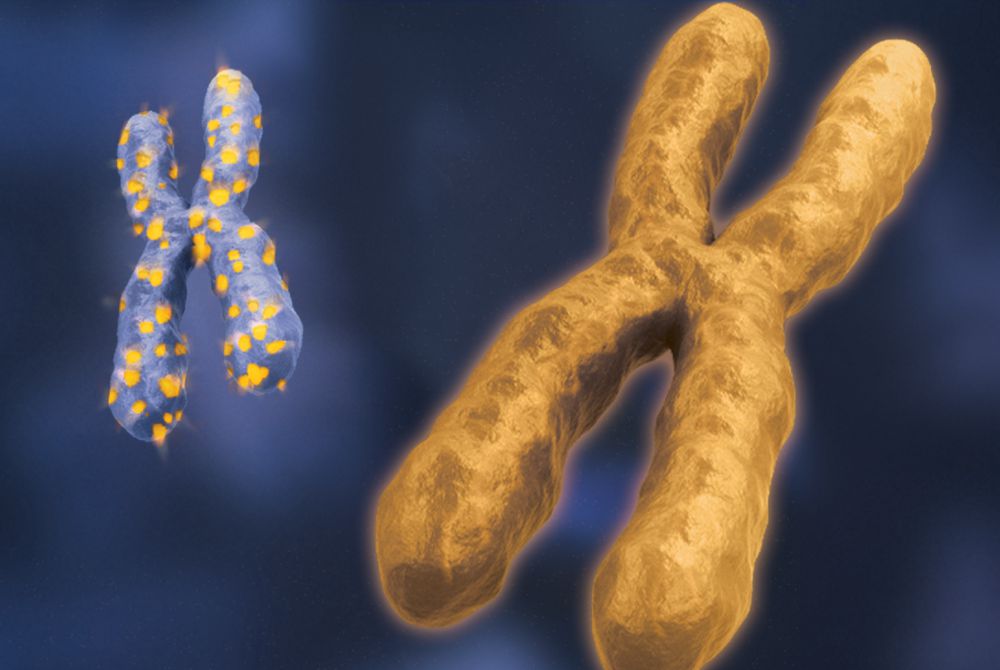
Мутация одной из Х-хромосом
Инструментальная диагностика
Среди инструментальных диагностических методов применяется лишь ЭЭГ (электроэнцефалография) и МРТ (магнитно-резонансная томография).
МРТ детально визуализирует структуры головного мозга. Томограф регистрирует уменьшенный объем мозга из-за малого количества белого вещества, уменьшенный размер среднего мозга, ответственного за слух, зрение, внимание и атрофию хвостатого ядра. Строение извилин остается нормальным.
ЭЭГ помогает выявить признаки эпилепсии по мозговой активности. При замедленном фоновом ритме диагноз подтверждается.
Чтобы выявить отклонения со стороны внутренних органов могут назначить:
- электрокардиографию (проверка состояния сердца);
- УЗИ органов брюшной полости;
- электронейромиографию (проверка нервной системы и мышц).
В 20-30% случаев выявляется физиологическая незрелость селезенки, печени или почек.
Дифференциальная диагностика
Проводят дифференциальную диагностику со схожим по симптоматике состоянием – аутизмом. В отличие от аутизма при синдроме Ретта имеются признаки прогредиентности (постепенное усиление симптоматики), но отсутствует искаженное психическое развитие, как при аутизме.
| Признаки | Характеристика синдрома Ретта | Характеристика аутизма |
| Неправильное физическое развитие у годовалых малышей | Не выявлено | Часто |
| Стереотипные движения | Однообразие стереотипий, наподобие «мытья рук» в зоне 1 пояса | Разнообразные движения верхними конечностями |
| Повторяющиеся манипуляции с предметами | Отсутствует | Присутствует: щелканье ручкой, размахивание перед собой каким-либо предметом, открывание/закрывание книги |
| Нарушенная координация (при поражении мозжечка) | Прогрессирование, обездвиженность | Поражения мозжечка отсутствует, но имеется манерность движений |
| Эпилептические припадки | Часто | Нет или совсем редко |
| Проблемы с дыхательной системой | Апноэ (временная продолжительная остановка дыхания) ночью либо днем | Отсутствуют |
| Нарушенный рост костей | Есть | Нет |
Симптомы синдрома Ретта напоминают другие состояния. Ошибочно за него могут принять состояние при дефиците орнитинтранскарбамилазы (сбой цикла мочевины) или восковидный липофусциноз нейронов, при котором прослеживается рефлекс наподобие «вязания спицами».
Подобные проявления судорожной атаксии, умственной отсталости встречаются также при синдроме Ангельмана.
Лечение
Учитывая генетическую природу синдрома Ретта, лечение еще не разработано. Имеются кое-какие наработки, проводятся испытания на мышах, однако уже есть обнадеживающие результаты. Получилось «излечить» пока 15% мутированных клеток мышей.
Проводились испытания на «бессмертных» клетках HeLa. При введении аминогликозидов (а именно гентамицина) восстанавливалась последовательность гена, претерпевшего несколько модификаций.
Основное лечение направлено на облегчение состояния больного. Применяется симптоматическая терапия, лечебная гимнастика, диетотерапия.
Медикаментозное лечение
Основной целью лечения является снизить выраженность симптомов посредством симптоматической терапии. Она включает прием:
- антиконвульсивных препаратов против судорог (Карбамазепин, Ламотриджин, вальпроевая кислота);
- снотворных (Мелатонин-С3, препараты барбитураты);
- антидепрессантов, противостоящих депрессивным состояниям (Сертралин, Амитриптилин);
- транквилизаторов, устраняющих тревожность (Клоназепам, Диазепам);
- антипсихотиков, воздействующих на психотические нарушения (Галоперидол, Хлорпромазин, Клозапин, Трифлуоперазин);
- ноотропов, улучшающих мозговую деятельность (Пирацетам, Актовегин, Пиритинол, Ноотропил). Ноотропы воздействуют на когнитивные процессы.
Если при приеме антипсихотиков, возникнут побочные экстрапирамидные расстройства, то показан прием м-холинолитиков (Тригексифенидила).
В профилактических целях назначаются препараты, улучшающие общие показатели здоровья. Чтобы предотвратить венозный застой, назначают препараты, улучшающие кровообращение. Это Инстенон, Фелодипин, Сермион.
Чтобы не образовывались тромбы, применяют антиагреганты (снижают склеивание клеток крови) – Аспирин. Также применяются антикоагулянты, регулирующие свертывающую систему крови – Гепарин.
Хирургического вмешательства не проводится.
Если психопатологическое расстройство пациента несет угрозу для него и окружающих или он сам неспособен к самообслуживанию и беспомощен, то больного принудительно госпитализируют в стационар.
Реабилитация
Излечить болезнь нельзя, но можно облегчить жизнь. Чтобы адаптировать пациента к социуму, проводят консультации с психологом, а чтобы исправить дефекты речи потребуются занятия с логопедом. Помимо этого, показана лечебная гимнастика или ЛФК.
С помощью лечебной физической культуры (ЛФК) можно предупредить искривление позвоночника, научить ребенка передвигаться. ЛФК хорошо сочетаема с лечебными массажами.
Улучшает психоэмоциональные показатели у детей Томатис-терапия (лечение музыкой). После прослушивания специально созданной музыки, пациенты изъявляют желание контактировать с другими людьми.
Адаптировать малыша к окружающей среде можно, используя еще несколько методов:
- гидрореабилитацию (терапия водными процедурами, минеральными ваннами, гидромассажем);
- игротерапию;
- арт-терапию (занятия искусством – рисованием или музыкой);
- иппотерапию (адаптивная верховая езда на лошади);
- фелинотерапию (общение с животными);
- дельфинотерапию.
Наилучшие результаты будут достигнуты, если реабилитация начнется раньше.
Питание
Педиатры предлагают диету с большим содержанием жиров, чтобы увеличить вес больного. Частое кормление маленькими порциями с интервалом в 3-4 часа стабилизирует состояние. Согласно данным французского Национального института здравоохранения, ребенку и подростку в день нужно съедать до 1300-1900 ккал, а взрослому, в зависимости от пола – от 1800 до 3000. Нужно учитывать это, подбирая диету ребенку. Чем больше выражаются стереотипии, судороги и раскачивания, тем больше калорий должен потреблять больной.
Как правило, у девочек, страдающих синдромом нет проблем с аппетитом, но процесс пережевывания и приема пищи обычно длительный. Занимает час или дольше.
Насыщенная жирами еда улучшает самочувствие, способствует набору веса и устраняет истощение. Применение кетогенной диеты, за основу которой берется преобладание жиров в блюдах, показало положительные результаты в борьбе с эпилептическими припадками, сопутствующими синдрому. Основа диеты это жиры, клетчатка, умеренное содержание белков и витамины.
Нельзя забывать о том, что у больных в 36% случаев имеются трудности с дыхательной системой, поэтому лучше измельчать некоторые продукты, особенно где есть волокна.
Важно измерять вес ребенка 1 раз в месяц, а если у него выставлен диагноз «истощение», то 1 раз в неделю. Недобор веса обусловлен недостаточным питанием или обезвоживанием. У девочек иногда отмечается повышенное слюноотделение, что также провоцирует потерю жидкости, поэтому пить воды нужно много.
У некоторых при употреблении жидкостей возникает поперхивание и кашель. Можно подавать более густые напитки, добавляя туда натуральные загустители. Во избежание кашля ребенок должен сидеть в положении, при котором подбородок будет касаться груди.
Правила кормления
При кормлении девочек с диагностированным синдромом следует придерживаться нескольких правил:
- если ребенок не способен самостоятельно держать вилку или ложку, необходимо имитировать его рукой поднесение пищи ко рту.
- подносить пищу стоит сбоку.
- блюда лучше давать по отдельности (сначала дать пережевать хлеб, а затем кашу);
- желательно приобщать малыша к процессу приготовления блюд, давать пробовать новые продукты, чтобы расширить его восприятие мира.
- можно давать размоченные в молоке ломтики печенья или хлеба для лучшего усваивания.
- страдающие синдромом привыкают к стереотипностям, поэтому желательно организовать их питание по времени.
Последствия и осложнения
На фоне сниженного интеллекта у детей появляются другие осложнения. Из-за слабости мышц появляется сколиоз. Впоследствии образовавшегося искривления позвоночника будут сдавливаться легкие, что спровоцирует их плохую вентиляцию.
Малоподвижный образ жизни и частое обезвоживание провоцируют запоры, способные привести к закупорке кишечника и невыносимым болям.
При трудностях сглатывания слюны и повышенном внутрибрюшном давлении существует риск желудочно-пищеводного рефлюкса (забрасывание содержимого желудка обратно в пищевод), что приведет к воспалению пищевода, респираторным инфекциям.

Высок риск утраты способности ходить.
Прогноз и продолжительность жизни
Прогноз не благоприятный, поскольку синдром гарантированно ведет к умственной отсталости тяжелой степени и прочим нарушениям. Больные синдромом могут дожить до 40-50 лет, при условии, что будет оказана поддерживающая терапия.
Причиной короткого срока жизни зачастую становится внезапная сердечная смерть. Помимо этого, около трети пациентов отмечаются пороки внутренних органов, что тоже становится неблагоприятным фактором. Высок риск дыхательной или полиорганной недостаточности, инсульта, некроза кишечника, отека головного мозга.
Профилактика
Выявить недуг можно лишь во время пренатальной (дородовой) диагностики с помощью генетических методов.
Первичную профилактику не проводят, поскольку предупредить генетический сбой нельзя. Вторичная профилактика (предупреждение лиц из группы риска) включает прием психофармакологических средств. Третичная профилактика направлена на заболевших и подразумевает предотвращение быстрого прогрессирования заболевания и преждевременной смерти. При третичной профилактике (комплаенс-терапии) проводится реабилитация с назначением препаратов-нейролептиков.
Заключение
Синдром Ретта – это заболевание, которое не подлежит излечению, но замедлить прогрессирование недуга можно лекарственными препаратами, гимнастикой и работой с психологом и логопедом. Как правило, продолжительность жизни короткая.


